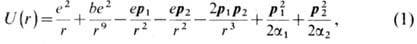
Химическая связь - межатомное взаимодействие, приводящее к образованию молекул или молекулярных
соединений. Химическая связь отличается от др. межатомных взаимодействий тем, что
при её возникновении происходит перестройка электронных оболочек связывающих
атомов. Химическая связь осуществляется либо путём перехода одного или неск. электронов
одного атома к другому (и о н н а я, или э л е кт р о в а л е н т н а я, X.
с.), либо обобществлением электронов парой (группой) атомов (к о в а л е н т
н а я, или гомео-полярная, X. с.). Устойчивость образующейся системы атомов
(молекулы) объясняется тем, что её энергия меньше суммарной энергии свободных
атомов; разность этих энергий наз. энергией X. с., она обычно ~200- 1000 кДж/моль
(2-10 эВ).
В образовании Химической связи участвуют
внешние, самые подвижные электроны атомов; электроны внутренних, полностью заполненных
электронных оболочек в этом процессе практически не участвуют, на их поведении
вступление атома в Химической связи сказывается слабо. X. с. обусловлена эл--магн. взаимодействием
атомов, однако в рамках классич. электродинамики достаточно точно описать X.
с. нельзя: молекула- квантовая система и подчиняется законам квантовой механики. Деление X. с. по механизму её осуществления на ионную и ковалентную условно,
т. к. реально в большинстве молекул X. с. носит смешанный характер и наз. с
е-м и п о л я р н о й X. с.
К Химической связи иногда относят донорно-акцепторную связь, а также металлическую связь, к-рые имеют энергию
связи того же порядка величины, что и X. с.
Ионная Химическая связь. характерна
для солей оснований и многих др. соединений, молекулы к-рых представляют собой
совокупность положительного и отрицательного ионов, связанных эл--статич. силами
притяжения. В 1916 В. Коссель (W. Kossel) предложил классич. теорию ионной связи,
к-рая объяснила мн. её особенности. В этой теории использовались нек-рые квантовые
представления о строении атома. Так, в ней постулировалось, что образование
ионов при ионной связи происходит т. о., чтобы ионы имели более устойчивую электронную
конфигурацию, близкую к конфигурации атомов инертных газов. Такая перестройка
должна быть связана с выделением энергии и; следовательно, с повышением устойчивости
системы.
Атомы металлов, имеющие
во внеш. электронной оболочке, как правило, один, два или три валентных электрона,
отдают их атому неметалла, у к-рого до заполнения внеш. электронной оболочки
не хватает одного, двух или трёх электронов. В таком процессе образуются два
иона с полностью заполненными внеш. электронными оболочками. Напр., при образовании
молекулы LiF атом Li с электронной конфигурацией 1s22s1
отдаёт электрон 2s атому F с электронной конфигурацией 1s22s2p5. Образующиеся ионы Li+ и F- имеют соответственно конфигурации
1s2 и 1s22s2p6, т. е. устойчивые электронные конфигурации атомов инертных газов Не и Ne
соответственно.
Прочность ионной X. с.
определяется потенц. энергией взаимодействия ионов U(r):
где е - заряд электрона;
r - расстояние между атомными ядрами; р1
и р2-дипольные моменты, образовавшиеся в
результате поляризации каждого иона в электрич. поле др. иона; a1
и a2 - их поляризуемости; b - эмпирич. константа. Первый член
учитывает энергию кулоновского притяжения ионов, второй - энергию обменного
отталкивания электронных оболочек (см. Обменное взаимодействие ),третий
и четвёртый члены характеризуют энергию взаимодействия свободных зарядов ионов
с диполями р1 и р2, пятый член описывает энергию взаимодействия диполей p1
и р2, шестой и седьмой - энергии деформации
диполей (в квазиупругом приближении). Равновесное межъядерное расстояние r0, при к-ром силы притяжения и отталкивания уравновешены, определяется из условия
dU/dr = 0; оно равно сумме радиусов ионов (см. Атомный радиус).
Ионные соединения в твёрдом
состоянии представляют собой ионные кристаллы. При испарении ионного соединения
из твёрдого состояния, расплава или раствора положит. и отрицат. ионы покидают
конденсированную фазу попарно, образуя в газообразном состоянии ионные молекулы.
Ионная X. с. возможна и при взаимодействии сложных (комплексных) ионов SO42-,
SiF62-, NH4+ и др., в к-рых атомы,
как правило, связаны ковалентной X. с.
Модель ионной Химической связи имеет ограниченную применимость. Она используется в теории внутрикристаллического
поля для объяснения свойств неорганич. координац. соединений, в к-рых центр.
ион переходного металла находится в поле окружающих его ионов или дипольных
молекул (в поле лигандов). В теории кристаллич. поля устойчивость координац.
соединения обеспечивается эл--статич. взаимодействием между центр. ионом и лигандами.
В поле лигандов уровни энергии центр. иона расщепляются (Штарка эффект), характер этого расщепления определяется симметрией поля лигандов.
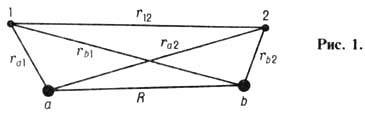
Ковалентная Химическая связь. возникает
при соединении в молекулу нейтральных атомов, валентные электроны к-рых обобществляются
участвующими в X. с. атомами. Этот тип Химической связи получил объяснение лишь в 1927
на основе квантовой механики, когда В. Гайтлер (W. Haitler) и Ф. Лондон (F.
London) построили квантовую теорию молекулы водорода. Молекула Н2
(рис. 1) состоит из двух ядер с зарядом
+ е (протонов)
а и b, находящихся друг от друга на расстоянии R, и двух
отрицательно заряженных электронов 1 и 2. Потенц. энергия взаимодействия этих
заряж. частиц между собой
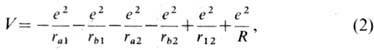
где первые 4 отрицат. члена
выражают энергию 1-го и 2-го электронов в поле своего и чужого ядра; пятый и
шестой (положительные) члены - энергию взаимного отталкивания электронов и отталкивания
ядер. Полная внутр. энергия молекулы (без учёта энергии её движения как целого
и влияния внеш. полей), кроме потенц. энергии взаимодействия составляющих её
частиц, включает и кинетич. энергию электронов и ядер. Полная энергия молекулы,
её осн. характеристика как квантовой системы, принимает дискретные значения
и определяет квантовые состояния молекулы. Значения 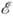 (R)полной внутр. энергии молекулы являются решением Шрёдингера уравнения
(R)полной внутр. энергии молекулы являются решением Шрёдингера уравнения
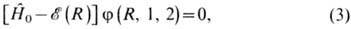
где оператор Гамильтона
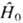 в т.
н. адиабатическом приближении (в пренебрежении движением ядер и спин-орбитальным
взаимодействием)можно записать в виде
в т.
н. адиабатическом приближении (в пренебрежении движением ядер и спин-орбитальным
взаимодействием)можно записать в виде
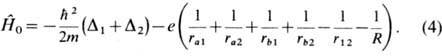
Для описания состояния
молекулы Н2 необходимо найти волновую функцию j(R, 1, 2) (здесь
1 и 2 - координаты электронов 1 и 2) этого состояния и энергию молекулы в нём.
Чтобы найти j(R, 1, 2), нужно решить ур-ние Шрёдингера в предположении,
что ядра находятся на достаточно большом фиксированном расстоянии R друг
от друга.
В нулевом приближении волновая
функция молекулы строится из волновых функций изолированных атомов ya
и yb. функция ya(1), учитывающая движение
1-го электрона в поле своего ядра, является решением ур-ния Шрёдингера для осн.
состояния атома Н с энергией 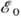 (13,6 эВ); то же самое можно сказать о функции yb (2). Полная
энергия молекулы в нулевом приближении, следовательно, равна 2
(13,6 эВ); то же самое можно сказать о функции yb (2). Полная
энергия молекулы в нулевом приближении, следовательно, равна 2 ,
а её волновая функция j, согласно Паули принципу ,должна быть антисимметричной
по отношению к перестановке пространств. и спиновых координат электронов. Поскольку
электроны принципиально неразличимы, безразлично, какой из них будет находиться
у определ. ядра. Линейная комбинация произведений ya(1)yb(2)
и ya(2)yb(1) позволяет построить два типа
антисимметричных координатных функций j, соответствующих синглетно-му (s)(спины
электронов антипараллельны) и триплет-ному (t)(спины параллельны) состояниям:
,
а её волновая функция j, согласно Паули принципу ,должна быть антисимметричной
по отношению к перестановке пространств. и спиновых координат электронов. Поскольку
электроны принципиально неразличимы, безразлично, какой из них будет находиться
у определ. ядра. Линейная комбинация произведений ya(1)yb(2)
и ya(2)yb(1) позволяет построить два типа
антисимметричных координатных функций j, соответствующих синглетно-му (s)(спины
электронов антипараллельны) и триплет-ному (t)(спины параллельны) состояниям:
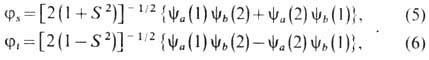
где
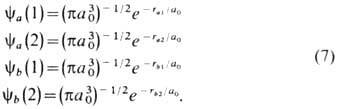
Здесь аа
=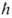 2/те2
- Бора радиус, т - масса электрона, е - его заряд,
2/те2
- Бора радиус, т - масса электрона, е - его заряд,
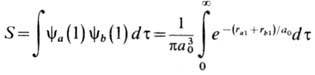
- интеграл перекрывания
волновых функций, dt - элемент объёма. При сближении атомов Н энергию
молекулы в первом приближении можно найти как ср. значение оператора в состоянии,
соответствующем волновым функциям нулевого приближения. Энергии системы в син-глетном
и триплетном состояниях можно записать в виде интегралов
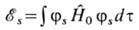 и
и 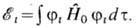
С учётом (5) и (6) получим
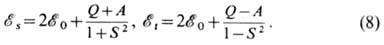
функция Q в (8) определяет
энергию кулоновского взаимодействия:
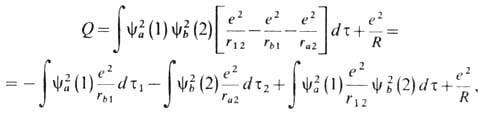
где первый интеграл определяет ср. значение энергии кулоновского притяжения электрона 1 к ядру b; второй - соответственно электрона 2 к ядру а (численно эти интегралы для молекулы Н2 равны); третий-энергию кулонов-ского отталкивания электронов; последний член соответствует энергии отталкивания ядер. функция А в (8) наз. обменным интегралом,
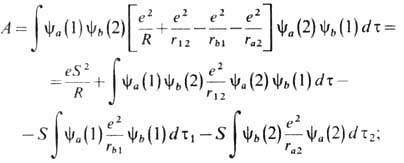
обменный интеграл отличен
от нуля только в тех точках пространства, где произведения ya(1)yb(1)
и ya (2)yb (2) отличны от нуля, т. е. в
области перекрывания электронных волновых функций атомов а и b. Значение
А экспоненциально убывает с расстоянием, поскольку волновые функции экспоненциально
убывают на больших расстояниях. Наличие обменной энергии А приводит к
тому, что в состоянии с антипараллельными спинами электронная плотность в пространстве
между ядрами увеличивается, а в состоянии с параллельными спинами - уменьшается,
т. е. сила отталкивания ядер возрастает. Т. о., разные свойства синглетного
и триплетного состояний молекулы количественно определяются значением обменного
интеграла А. На рис. 2 приведены зависимости 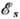 и
и 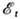 от
межъядерного расстояния
R. Образование Химической связи происходит только при
от
межъядерного расстояния
R. Образование Химической связи происходит только при 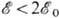 , т. е. в состоянии с энергией
, т. е. в состоянии с энергией 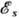 .
Теория Гайтлера- Лондона для равновесного радиуса молекулы Н2, соответствующего
её стабильному состоянию, даёт значение R0 = 8,7 нм,
эксперим. значение R0 = 7,4 нм. Значительно лучшее
согласие теоретич. и эксперим. значений для R0 можно
получить на основе вариац. методов расчёта.
.
Теория Гайтлера- Лондона для равновесного радиуса молекулы Н2, соответствующего
её стабильному состоянию, даёт значение R0 = 8,7 нм,
эксперим. значение R0 = 7,4 нм. Значительно лучшее
согласие теоретич. и эксперим. значений для R0 можно
получить на основе вариац. методов расчёта.
Непосредств. взаимодействие
спинов электронов в молекуле Н2 практически не играет роли в образовании
X. с. Энергия такого взаимодействия меньше обменной энергии. Кроме того, устойчивая
X. с. образуется и в молекулярном ионе H+2, состоящем
из двух ядер и только одного электрона, т. е. в отсутствие обменного взаимодействия.
Т. о., для объяснения X. с. достаточно рассмотреть лишь силы притяжения и отталкивания
между электронами, не вводя "обменных" сил. Обменный интеграл А появляется только в случае приближённого решения задачи; при точном решении
задачи из энергии нельзя выделить её обменную часть.
Для сложных молекул количеств.
теория ковалентной Х.с. не построена из-за непреодолимых матем. трудностей.
Поэтому вводятся модельные представления, к-рые, опираясь на теорию молекулы
водорода, позволяют качественно объяснить особенности ковалентных взаимодействий
в сложных молекулах. Напр., на основе представления о спаренных электронах можно
объяснить свойство насыщения ковалентных связей. Каждая ковалентная связь образуется
при спаривании их валентных электронов. В квантовой химии спаренными
наз. электроны, занимающие одно и то же координатное состояние (напр., в атоме
Не оба электрона находятся в состоянии 1s), но спины их имеют
противоположное направление. Третий электрон, согласно принципу Паули, уже не
может находиться в таком же координатном состоянии. Поэтому взаимодействие любой
спаренной пары электронов атома или молекулы с электронами др. атома приводит
к отталкиванию. Два электрона молекулы Н2, образующие ковалентную
связь, в синглетном спиновом состоянии также являются спаренными, а Х.с. между
ними - насыщенной.
Свойства молекулы определ.
не только её составом, но и пространств. расположением атомов в ней. Хим. валентности
обладают определ. направленностью в том случае, когда валентных электронов два
и более. Так, у атома N в осн. состоянии 1s22s22p2
электроны 1s и 2s спарены и не участвуют в Х.с., а три валентных
2р-электрона находятся в таких координатных состояниях, что направления,
в к-рых плотность вероятности пространств. распределения электронов максимальна,
образуют углы 90°. X. с., образуемые при участии этих валентных электронов
атома N, также должны составлять прямые углы, т. к. при сближении атомов в этих
направлениях волновые функции электронов перекрываются наиб. сильно. Молекула
NH3 действительно имеет пирамидальное строение, однако углы между
связями N - Н составляют не 90°, а 107° 18', что является следствием
взаимного отталкивания ядер водорода, лежащих в основании пирамиды.
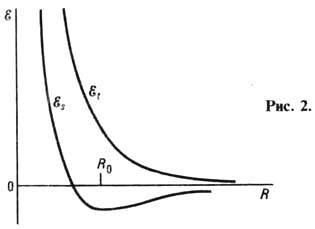
По симметрии распределения
электронной плотности вдоль линии связи различают s-, p-, d- и т.д. связи. При
s-связи (рис. 3) макс. перекрывание электронных функций наблюдается вдоль линии
связи - электронные функции перекрываются одним лепестком и оба атома могут вращаться
вокруг оси, совпадающей с линией связи. p-связь менее прочна, чем s-связь, перекрывание
волновых функций происходит в два лепестка, поворот атомов вокруг линии связи
исключён. При d-связях происходит перекрывание волновых функций в 4 лепестка с
каждой стороны. Между двумя атомами возможна только одна s-связь, но наряду
с ней между теми же атомами могут быть две p-связи.
Существуют молекулы, в
к-рых нельзя выделить отд. Х.с., попарно соединяющие соседние атомы. В таких
соединениях электроны рассредоточены (делокализованы) вдоль всей системы атомов,
а X. с. носит невалентный "орбитальный" характер. В этом случае
Х.с. нельзя описать с помощью одноэлектронных волновых функций и Х.с. рассчитывается
на основе вариац. методов.
Х.с. изображают разл. способами.
Ионные соединения часто записывают в хим. символах элементов со знаками зарядов
(Li+ F-; K+Br-). Органич. катионы
и анионы заключают в скобки ([R4N]+Сl-). Простую
ковалентную Х.с. изображают двумя точками или чертой между хим. символами (Н:Сl
или Н - Сl). Два электрона-по одному от каждого атома - наз. п о д е л ё н н
о й п а р о й. Если X.с. осуществляется тремя поделёнными парами (напр., в молекуле
N2), то её изображают тремя чёрточками (N = N). В молекуле
могут присутствовать электронные пары, не участвующие в Х.с. и принадлежащие
одному атому, они наз. неподелёнными парами, их изображают в структурных ф-лах
в виде двух точек или чёрточек, напр.
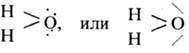
Неподелённые пары могут
осуществить X. с. при условии, что у вступающего в неё др. атома не заполнена
внеш. электронная оболочка (электронный "пробел"). Такого рода связь
является разновидностью ковалентной связи и наз. координационной Х.с. (или донорно-акцепторной
связью). Она изображается стрелкой, направленной в сторону отрицательно заряженного
атома (акцептора), к-рую иногда заменяют простой чертой, обозначая при этом
знаки заряда у атомов:

Методы исследования
Х.с. предполагают сочетание теории с экспериментом. В совр. теоретич. расчётах
используют формализм матрицы плотности, позволяющий характеризовать одночастичные
состояния для систем, содержащих неск. разных или тождественных частиц.
Характер X. с. влияет на мн. свойства вещества, исследование к-рых позволяет получить информацию о X. с. К экс-перим. методам изучения X. с. относятся разл. виды спектроскопии (см., напр., Инфракрасная спектроскопия, Молекулярные спектры, Спектры кристаллов и др.), дифракционные методы (см. Рентгеновский структурный анализ, Электронография, Нейтронография), магнетохи-мия, химическая кинетика, резонансные методы (ЭПР, ЯМР) и др.

Л. Ф. Уткина.
|
|
