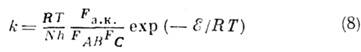Химическая кинетика - область физ. химии, в к-рой изучают механизмы и скорости хим. реакций.
Химическая кинетика включает три осн. задачи: изучение закономерностей протекания хим. реакций
во времени и зависимость их скоростей от концентраций реагентов, температуры и др.
факторов; теоретич. определение констант скоростей хим. реакций на основе молекулярного
строения реагентов; исследование хим. реакций в условиях движения вещества,
диффузии реагентов, наличия теплопередачи и т. д. (хим. макрокинетика).
Основные понятия и законы химической кинетики
В процессе хим. реакции могут происходить как однотипные элементарные
акты (простые, одностадийные реакции, напр. Н2+I2=2НI),
так и элементарные акты разл. типа, число к-рых может достигать мн. десятков
и сотен (сложные, многостадийные реакции, напр, образование молекул Н2O идёт через элементарные реакции типа H2;
,
.
Стехиометрич. ур-ние сложной реакции, включающее только исходные и конечные
вещества (напр., 2Н2+О2=2Н2О), не описывает
механизма происходящих процессов. При сложных реакциях в нек-рых стадиях возникают
промежуточные, лабильные продукты (в рассмотренном примере - Н, О и ОН), концентрация
к-рых обычно невелика, т. к. они быстро потребляются в др. стадиях. Под механизмом
хим. реакции понимается совокупность элементарных стадий, составляющих процесс
превращения исходных веществ в конечные продукты.
Скорость к--л. стадии хим.
реакции равна числу элементарных актов данного типа в единицу времени в единице
объёма (для гомогенных реакций, протекающих в объёме одной фазы) или на единице
поверхности (для гетерогенных реакций, протекающих на поверхности раздела фаз).
Наиб. развита К. х. гомогенных реакций в газовой фазе, т. к. одна из её основ
- хорошо разработанная кинетическая теория газов.
Скорость хим. реакции зависит
от температуры и давления, а при заданных внеш. условиях является функцией концентраций
реагирующих веществ. В идеальных газовых
смесях и идеальных (разбавленных) растворах скорость
простой, одностадийной реакции подчиняется закону действующих масс. Если хим.
реакция обратима:
(Аi и
Вj - символы реагирующих веществ, аi и bj - стехиометрич. коэффициенты, n и m - числа реагирующих веществ), то, согласно
закону действующих масс, скорость прямой (и обратной) реакций пропорц. произведению
концентраций реагирующих веществ, взятых в степенях, равных соответствующим
стехиометрич. коэффициентам. Обозначая через [А] и [В]
концентрацию веществ А и В для скоростей прямой
и обратной
реакций, получим
Коэф. k+ и k_ наз. константами скоростей хим. реакций.
При равенстве скоростей
прямой и обратной реакций наступает хим. равновесие, при к-ром
где К - константа химического равновесия.
Физ. интерпретация закона
действующих масс достаточно проста. Элементарный хим. акт происходит лишь в
том случае, когда a1 частиц типа А1, а2
частиц типа А2 и т. д. сблизятся на расстояние порядка размеров
молекул. Вероятность же такой встречи пропорц. концентрациям реагирующих веществ,
взятых в степенях, равных соответствующим стехиометрич. коэффициентам.
При обычных условиях вероятность
одноврем. встре-чи более чем трёх частиц крайне мала, поэтому наблюдаются лишь
элементарные акты, включающие в себя распад отд. молекулы или реакции между
двумя или тремя частицами. Они наз. соответственно мономолекулярными, бимолекулярными
и тримолекулярными реакциями. Сумма стехиометрич. коэффициентов исходных веществ
или число молекул, участвующих в элементарном акте, наз. порядком реакции, к-рый
для простых реакций не превышает трёх. Порядок реакции по данному веществу равен
его стехиометрич. коэффициенту.
Зависимость концентраций
реагентов и продуктов простой реакции от времени получается интегрированием
кинетич. ур-ния. Скорость реакции по определению равна
Учитывая нач. условия t=0,
[Аi]=[Ai]0, [Вj]=[Вj]0,
можно выразить концентрации всех веществ через концентрацию одного из них, напр.
A1, и из закона действующих масс (2) и соотношений (4) получить
кинетич. ур-ние для [A1]:
причём функция F кроме
заданных нач. концентраций зависит только от одной переменной - концентрации
вещества А1, Интегрируя (5) по времени, можно получить [A1(t)],
а следовательно, [Ai(t)] и [Bj(t)]. График
зависимости концентрации вещества, участвующего в реакции, от времени называется
кинетической кривой.
Для сложных, многостадийных
реакций закон действующих масс выполняется лишь для отд. стадий, но не для стехиометрич.
ур-ния реакции. Исследование кинетики
таких реакций проводится на основе системы кинетич. ур-ний:
к-рые представляют собой
законы сохранения для всех участвующих в реакции веществ. функции Fi
, представляют собой сумму выражений типа (2), каждое из слагаемых к-рых является
вкладом к--л. элементарного акта в образование или расход компонента Xi.
Число нелинейных ур-ний в системе (6) в общем случае велико, и интегрирование
её возможно чаще всего лишь при использовании быстродействующих ЭВМ. Возникающие
при этом трудности связаны с разбросом в неск. порядков значений констант скоростей
элементарных стадий реакции. Иногда скорость сложной хим. реакции записывают
в виде "эффективного" закона действующих масс для стехиометрич.
ур-ния, т. е. через концентрации исходных веществ.
В связи с огромным многообразием
сложных хим. реакций полная их классификация вряд ли возможна. Реакции типа
и ,
наз.
соответственно последовательными и параллельными. Реакции, протекающие в присутствии
катализатора, т. е. вещества, к-рое вызывает или ускоряет реакцию, но не расходуется
в её ходе, наз. каталитическими (автокаталитическими, если катализ осуществляется
промежуточными или конечными продуктами). Реакция, идущая под влиянием другой
реакции, наз. индуцированной или сопряжённой.
Важное место среди сложных
реакций занимают цепные реакции, в к-рых один первичный акт активации приводит
к превращению большого числа молекул исходных веществ. Цепная реакция начинается
с акта зарождения цепи, в к-ром из молекул исходных веществ образуются активные
частицы - атомы и радикалы, высокая реакционная способность к-рых связана с
наличием у них одного или неск. неспаренных электронов. В результате взаимодействия
таких активных частиц с молекулами вновь появляются новые атомы и радикалы.
Если в этой стадии цепной реакции, наз. продолжением цепи, число неспаренных
электронов не меняется, то реакция наз. неразветвлённой, в противном случае
говорят о разветвлённой цепной реакции. Так, неразветвлённая цепная реакция
хлорирования водорода, стехиометрич. ур-ние к-рой Н2+С12=2НС1,
содержит три элементарные стадии: 1) С122С1,
2) С1+ +Н2НС1+Н,
3) Н+С12НС1+С1.
Наиб. изученная разветвлённая цепная реакция - реакция образования воды, к-рая
протекает по стехиометрич. ур-нию 2Н2+ +О2=2Н2О,
её осн. элементарные стадии: 1) Н2+О2
Н+НО2, 2) Н2+ОН+ОН,
3) ОН+Н2Н2О+Н,
4) О2+НОН+О.
В приведённых примерах активными частицами являются атомы С1, О и Н и радикал
гидроксила ОН. Реакции типа 1) наз. реакциями зарождения цепи - из насыщенных
молекул возникают активные частицы. Реакции 2) и 3) - продолжение цепи: из одних
активных частиц получаются активные частицы др. типа. Реакция 4) - разветвление
цепи - число неспаренных электронов меняется от 1 до 3 (у атома Н и гидроксила
ОН - по одному неспаренному электрону, у атома О - два).
В процессе развития цепной
реакции может происходить гибель атомов и радикалов на стенках реакционного
сосуда или в объёме реагирующей среды. Конкуренция гибели и размножения их в
разветвлённых цепных реакциях приводит к своеобразным предельным явлениям (см.
Взрыв).
Константы скоростей химической реакции
В задачу К. х. входит теоретич. определенно констант
скоростей элементарных стадий. Они зависят от температуры Т и типа реагирующих
молекул. Температурная зависимость скорости реакции определяется законом Арре-ниуса:
где
- предэкспоненциальный множитель, слабо зависящий от Т,
- энергия активации, R - универсальная газовая постоянная. Закон
Аррениуса свидетельствует о том, что для вступления в реакцию реагенты должны
преодолеть потенц. барьер, высота к-рого
, а множитель
, согласно распределению Максвелла - Больцмана (см. Болъцмана распределение), пропорционален доле реагирующих частиц, имеющих энергию теплового движения,
большую высоты барьера. Типичные значения энергии активации
десятки ккал/моль. При реакциях активных атомов и радикалов энергия активации
меньше и, в частности, может равняться нулю.
Кинетич. теория газов позволяет
дать оценку пред-экспонентального множителя z для би- и тримолеку-лярных реакций
в газовой фазе. Скорость бимолекулярной реакции А+В пропорц. кол-ву двойных
соударений z0[A][B], причём фактор двойных столкновений
где rA ,
rB и МА, МB - радиусы и массы молекул.
Т. к. хим. реакция может произойти только при определ. ориентации молекул в
момент столкновения, то k = pz0, где р - вероятность
благоприятной ориентации, наз. стерич. фактором. Аналогичным образом может быть
оценена и константа скорости тримолекулярной реакции. Поскольку р и z
меньше единицы, факторы столкновений дают макс, оценку для констант скоростей
реакции (для бимолекулярной реакции z0~10-10-
10-11 см3/с, для тримолекулярной реакции z0~
10-33-
10-35 см3/с).
При мономолекулярных реакциях
превращению подвергаются отд. частицы, обладающие избыточной энергией. Распад
молекулы происходит при концентрации этой энергии на определ. хим. связи, к-рая
в момент реакции разрывается. Если
- необходимая для разрыва связи энергия, а
- частота внутримолекулярных колебаний, то
. Типичные значения констант скоростей мономолекулярных реакций 1013-1014
с-1.
В жидкой фазе каждая молекула
в течение определ. времени (10-9-10-10 с) совершает колебания
в окружении ближайших соседей, а затем перескакивает в др. положение равновесия.
Если произошёл распад молекулы, то образовавшаяся пара радикалов находится в
непосредств. близости друг от друга. Рекомбинация радикалов уменьшает константу
скорости реакций в жидкой фазе по сравнению с реакциями в газе.
Нахождение абс. значения
константы скорости хим. реакции через характеристики реагирующих молекул - задача
квантовой химии. Её решение наталкивается на значит. трудности в связи
с большим числом участвующих в реакции частиц (ядер, электронов). Поскольку
при реакции происходит перегруппировка атомов, то меняется и потенц. энергия
системы, зависящая от координат ядер атомов. На первом этапе решения задачи
находится рельеф многомерной потенц. энергии с целью найти наиб. выгодный путь
реакции, при к-ром реагирующая система преодолевает энергетич. барьер мин. высоты.
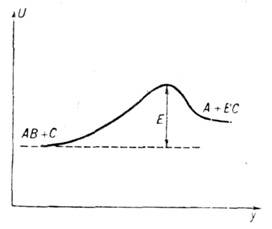
Потенц. поверхность для реакции АВ+С
А+СВ (все атомы находятся на одной прямой) состоит из двух "долин"
1 и 2, разделённых "перевалом" 3 (рис.
1). Энергетически наивыгоднейший путь обозначен пунктиром - он проходят через
перевальную точку. Разрез потенц. поверхности вдоль реакц. пути изображён на
рис. 2. Подавляющее большинство реально осуществляющихся элементарных
актов развиваются по путям, близким к проходящему через перевальную точку. Состояние
системы атомов, находящейся в пере- -вальной точке или в непосредственной близости
от неё, наз. переходным состоянием или активированным комплексом. Введение состояния,
переходного между исходным и конечным состояниями, позволяет применить для вычисления
константы скорости реакции методы статистич. механики.
Рис. 1. Поверхность
потенциальной энергии для реакции АВ+ + СА+ВС
[проекция уровней одинаковой энергии на плоскость (rAB, rBC,),r
- расстояние между атомами].
Рис. 2. Изменение
потенциальной энергии U вдоль реакционного пути (у - координата
реакции).
В методе переходного состояния,
или активированного комплекса, предполагается, что равновесное распределение
Максвелла-Больцмана не нарушается, акт реакции протекает адиабатически (электроны
движутся гораздо быстрее ядер), движение ядер можно рассматривать методами классич.
механики. Эти предположения позволяют найти концентрацию активированных комплексов
и скорость их перехода через критич. конфигурацию, а следовательно, константу
скорости хим. реакции. Последняя выражается через статистические суммы исходных
частиц FAB , FC активированного комплекса Fa.k.
. Так, для рассмотренной выше бимолекулярной реакции
(N - число Авогадро,
h - Планка постоянная). При развитии теории скорости хим. реакции необходимо
в нек-рых случаях учитывать искажения равновесного распределения за счёт самой
хим. реакции и возможность подбарьерного прохождения частиц (туннельный эффект).
Макрокинетика химических
реакций. Во мн. случаях (особенно в процессах хим. технологии) хим. превращение
происходит в условиях, осложнённых разл. физ. факторами (выделение тепла и его
отвод, движение вещества, перемешивание смеси, диффузия реагентов, подвод реагентов
и удаление продуктов из реакц. сосуда). Учёт этих факторов - задача м а к р
о с к о п и ч. хим. кинетики. Характеристики элементарного хим. акта, взятые
из микроскопич. теории или эксперимента, вводятся в ур-ния механики сплошных
сред (ур-ния теплопроводности, диффузии, гидродинамики), решение к-рых позволяет
рассчитать течение хим. превращения в реальных ситуациях.
Литература по химической кинетике
Франк-Каменецкий Д.А., Диффузия и теплопередача в химической кинетике, 3 изд., М., 1987:
Глесстон C., Лейдлер К., Эйринг Г., Теория абсолютных скоростей реакций, пер. с англ., М., 1948:
Семенов Н. Н., О некоторых проблемах химической кинетики и реакционной способности. 2 изд., М., 1958:
Эмануэль Н. М., Кнорре Д. Г., Курс химической кинетики, 4 изд., М., 1984;
Бенсон С., Основы химической кинетики, пер. с англ., М., 1964;
Денисов Е. Т., Кинетика гомогенных химических реакций, М., 1978;
Кондратьев В. Н., Ни-китин Е. Е., Химические процессы в газах, М. 1981.
Знаете ли Вы, что такое "Большой Взрыв"? Согласно рупору релятивистской идеологии Википедии "Большой взрыв (англ. Big Bang) - это космологическая модель, описывающая раннее развитие Вселенной, а именно - начало расширения Вселенной, перед которым Вселенная находилась в сингулярном состоянии. Обычно сейчас автоматически сочетают теорию Большого взрыва и модель горячей Вселенной, но эти концепции независимы и исторически существовало также представление о холодной начальной Вселенной вблизи Большого взрыва. Именно сочетание теории Большого взрыва с теорией горячей Вселенной, подкрепляемое существованием реликтового излучения..." В этой тираде количество нонсенсов (бессмыслиц) больше, чем количество предложений, иначе просто трудно запутать сознание обывателя до такой степени, чтобы он поверил в эту ахинею. На самом деле взорваться что-либо может только в уже имеющемся пространстве. Без этого никакого взрыва в принципе быть не может, так как "взрыв" - понятие, применимое только внутри уже имеющегося пространства. А раз так, то есть, если пространство вселенной уже было до БВ, то БВ не может быть началом Вселенной в принципе. Это во-первых. Во-вторых, Вселенная - это не обычный конечный объект с границами, это сама бесконечность во времени и пространстве. У нее нет начала и конца, а также пространственных границ уже по ее определению: она есть всё (потому и называется Вселенной). В третьих, фраза "представление о холодной начальной Вселенной вблизи Большого взрыва" тоже есть сплошной нонсенс. Что могло быть "вблизи Большого взрыва", если самой Вселенной там еще не было? Подробнее читайте в FAQ по эфирной физике.
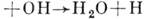 ;
;
 ,
,
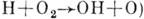 .
Стехиометрич. ур-ние сложной реакции, включающее только исходные и конечные
вещества (напр., 2Н2+О2=2Н2О), не описывает
механизма происходящих процессов. При сложных реакциях в нек-рых стадиях возникают
промежуточные, лабильные продукты (в рассмотренном примере - Н, О и ОН), концентрация
к-рых обычно невелика, т. к. они быстро потребляются в др. стадиях. Под механизмом
хим. реакции понимается совокупность элементарных стадий, составляющих процесс
превращения исходных веществ в конечные продукты.
.
Стехиометрич. ур-ние сложной реакции, включающее только исходные и конечные
вещества (напр., 2Н2+О2=2Н2О), не описывает
механизма происходящих процессов. При сложных реакциях в нек-рых стадиях возникают
промежуточные, лабильные продукты (в рассмотренном примере - Н, О и ОН), концентрация
к-рых обычно невелика, т. к. они быстро потребляются в др. стадиях. Под механизмом
хим. реакции понимается совокупность элементарных стадий, составляющих процесс
превращения исходных веществ в конечные продукты.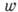 простой, одностадийной реакции подчиняется закону действующих масс. Если хим.
реакция обратима:
простой, одностадийной реакции подчиняется закону действующих масс. Если хим.
реакция обратима: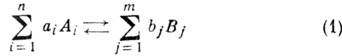
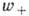 и обратной
и обратной 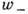 реакций, получим
реакций, получим
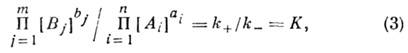
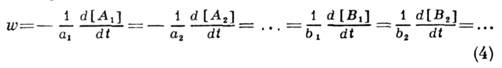

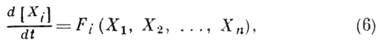
 и
и 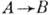 ,
,
 наз.
соответственно последовательными и параллельными. Реакции, протекающие в присутствии
катализатора, т. е. вещества, к-рое вызывает или ускоряет реакцию, но не расходуется
в её ходе, наз. каталитическими (автокаталитическими, если катализ осуществляется
промежуточными или конечными продуктами). Реакция, идущая под влиянием другой
реакции, наз. индуцированной или сопряжённой.
наз.
соответственно последовательными и параллельными. Реакции, протекающие в присутствии
катализатора, т. е. вещества, к-рое вызывает или ускоряет реакцию, но не расходуется
в её ходе, наз. каталитическими (автокаталитическими, если катализ осуществляется
промежуточными или конечными продуктами). Реакция, идущая под влиянием другой
реакции, наз. индуцированной или сопряжённой. 2С1,
2) С1+ +Н2
2С1,
2) С1+ +Н2 НС1+Н,
3) Н+С12
НС1+Н,
3) Н+С12 НС1+С1.
Наиб. изученная разветвлённая цепная реакция - реакция образования воды, к-рая
протекает по стехиометрич. ур-нию 2Н2+ +О2=2Н2О,
её осн. элементарные стадии: 1) Н2+О2
НС1+С1.
Наиб. изученная разветвлённая цепная реакция - реакция образования воды, к-рая
протекает по стехиометрич. ур-нию 2Н2+ +О2=2Н2О,
её осн. элементарные стадии: 1) Н2+О2 Н+НО2, 2) Н2+О
Н+НО2, 2) Н2+О Н+ОН,
3) ОН+Н2
Н+ОН,
3) ОН+Н2 Н2О+Н,
4) О2+Н
Н2О+Н,
4) О2+Н ОН+О.
В приведённых примерах активными частицами являются атомы С1, О и Н и радикал
гидроксила ОН. Реакции типа 1) наз. реакциями зарождения цепи - из насыщенных
молекул возникают активные частицы. Реакции 2) и 3) - продолжение цепи: из одних
активных частиц получаются активные частицы др. типа. Реакция 4) - разветвление
цепи - число неспаренных электронов меняется от 1 до 3 (у атома Н и гидроксила
ОН - по одному неспаренному электрону, у атома О - два).
ОН+О.
В приведённых примерах активными частицами являются атомы С1, О и Н и радикал
гидроксила ОН. Реакции типа 1) наз. реакциями зарождения цепи - из насыщенных
молекул возникают активные частицы. Реакции 2) и 3) - продолжение цепи: из одних
активных частиц получаются активные частицы др. типа. Реакция 4) - разветвление
цепи - число неспаренных электронов меняется от 1 до 3 (у атома Н и гидроксила
ОН - по одному неспаренному электрону, у атома О - два).
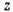 - предэкспоненциальный множитель, слабо зависящий от Т,
- предэкспоненциальный множитель, слабо зависящий от Т, 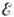 - энергия активации, R - универсальная
- энергия активации, R - универсальная 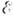 , а множитель
, а множитель 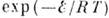 , согласно
, согласно 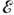 десятки ккал/моль. При реакциях активных атомов и радикалов энергия активации
меньше и, в частности, может равняться нулю.
десятки ккал/моль. При реакциях активных атомов и радикалов энергия активации
меньше и, в частности, может равняться нулю.
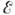 - необходимая для разрыва связи энергия, а
- необходимая для разрыва связи энергия, а  - частота внутримолекулярных
- частота внутримолекулярных 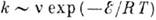 . Типичные значения констант скоростей мономолекулярных реакций 1013-1014
с-1.
. Типичные значения констант скоростей мономолекулярных реакций 1013-1014
с-1. А+СВ (все атомы находятся на одной прямой) состоит из двух "долин"
1 и 2, разделённых "перевалом" 3 (рис.
1). Энергетически наивыгоднейший путь обозначен пунктиром - он проходят через
перевальную точку. Разрез потенц. поверхности вдоль реакц. пути изображён на
рис. 2. Подавляющее большинство реально осуществляющихся элементарных
актов развиваются по путям, близким к проходящему через перевальную точку. Состояние
системы атомов, находящейся в пере- -вальной точке или в непосредственной близости
от неё, наз. переходным состоянием или активированным комплексом. Введение состояния,
переходного между исходным и конечным состояниями, позволяет применить для вычисления
константы скорости реакции методы статистич. механики.
А+СВ (все атомы находятся на одной прямой) состоит из двух "долин"
1 и 2, разделённых "перевалом" 3 (рис.
1). Энергетически наивыгоднейший путь обозначен пунктиром - он проходят через
перевальную точку. Разрез потенц. поверхности вдоль реакц. пути изображён на
рис. 2. Подавляющее большинство реально осуществляющихся элементарных
актов развиваются по путям, близким к проходящему через перевальную точку. Состояние
системы атомов, находящейся в пере- -вальной точке или в непосредственной близости
от неё, наз. переходным состоянием или активированным комплексом. Введение состояния,
переходного между исходным и конечным состояниями, позволяет применить для вычисления
константы скорости реакции методы статистич. механики.
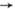 А+ВС
[проекция уровней одинаковой энергии на плоскость (rAB, rBC,),r
- расстояние между атомами].
А+ВС
[проекция уровней одинаковой энергии на плоскость (rAB, rBC,),r
- расстояние между атомами].